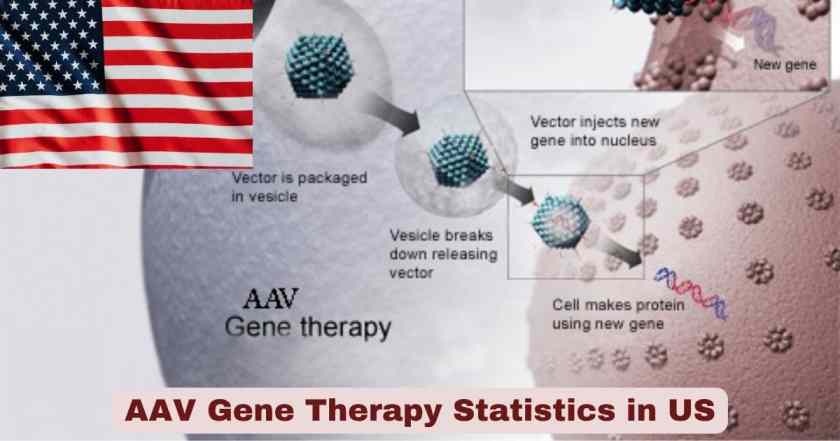
What Is AAV Gene Therapy?
Adeno-associated virus (AAV) gene therapy is the most clinically advanced and commercially proven platform for delivering functional genetic material directly into human cells in vivo — inside a living patient — to treat or cure diseases caused by faulty or missing genes. An AAV is a small, naturally occurring virus roughly 25 nanometers in diameter that is non-pathogenic, meaning it does not cause disease in humans under normal circumstances. Researchers remove its own viral genes and replace them with a therapeutic transgene — a corrected or replacement copy of the defective gene responsible for the patient’s condition — along with regulatory sequences that control where and how strongly the gene is expressed. This engineered “recombinant AAV” (rAAV) particle then acts as a biological delivery vehicle, entering cells, crossing into the nucleus, and expressing the therapeutic protein for months or years. AAV’s unique properties — broad tissue tropism across more than 12 naturally occurring serotypes (AAV1 through AAV12), favorable safety profile, largely non-integrating character, ability to cross the blood-brain barrier (AAV9 specifically), and long-term stable gene expression — have made it the most widely used viral vector in gene therapy clinical trials worldwide. As of 2025, 7 AAV-based gene therapy products have received regulatory approval globally from either the FDA or EMA, and the clinical pipeline spans hundreds of active trials across neurology, hematology, ophthalmology, muscle diseases, and metabolism.
What makes the AAV gene therapy landscape in 2025–2026 particularly significant — and genuinely complex — is that the field is simultaneously experiencing its greatest commercial milestones and its most serious scientific and commercial recalibration. The global AAV gene therapy market was valued at approximately $2.75–3.85 billion in 2024–2025 (depending on scope and methodology) and is projected to reach $3.7–5.4 billion in 2026, with long-term projections ranging from $23 billion to $112 billion by 2034–2035 at CAGRs of 26–40%. North America dominates with 42–54% of global market share, reflecting the US’s leadership in gene therapy R&D, regulatory infrastructure, and commercial launch environment. Yet 2025 also brought significant setbacks: Sarepta Therapeutics reported three patient deaths linked to AAV gene therapies for Duchenne muscular dystrophy, triggering clinical holds; Biogen discontinued all AAV-based gene therapy programs in September 2025; and Vertex Pharmaceuticals exited AAV research entirely in April 2025. These are not signs that the field is failing — they are signs that it is maturing, moving from early proof-of-concept into the harder questions of long-term durability, manufacturing scalability, immunogenicity management, and sustainable pricing that determine whether transformative science becomes accessible medicine.
Interesting Facts about AAV Gene Therapy
| Fact | Detail |
|---|---|
| AAV full name | Adeno-Associated Virus — a small, non-pathogenic single-stranded DNA virus |
| AAV particle size | ~25 nanometers in diameter |
| AAV naturally occurring serotypes | 12 naturally occurring serotypes (AAV1–AAV12) + over 100 variations |
| Most clinically used serotype | AAV9 — crosses blood-brain barrier; used in Zolgensma; 43% market share by serotype |
| AAV genome packaging limit | ~4.7–4.8 kb of recombinant DNA — a key limitation for large genes |
| Global AAV gene therapy market (2024) | $2.75 billion across top 7 markets (US, EU4, UK, Japan) — IMARC Group |
| Global AAV gene therapy market (2025) | $2.85–3.85 billion (range across research firms) |
| Global AAV gene therapy market (2026) | $3.74–5.40 billion (range across research firms) |
| Market CAGR (2025–2034/2035) | 26.43–40.1% depending on firm and timeframe |
| Long-term market projection (2034/2035) | $23.5–112.2 billion depending on scope |
| North America market share | 42–54% of global AAV gene therapy market |
| AAV9 CDMO market share (2025) | 40.92% — largest serotype in CDMO market |
| FDA-approved AAV gene therapy products (as of 2025) | 7 AAV-based products globally (FDA + EMA) |
| Global cell and gene therapy clinical trials (H1 2025) | 1,905 ongoing clinical trials — ARM/ASGCT data |
| North America CGT clinical trials (H1 2025) | 844 active trials in North America |
| AAV clinical trial growth rate | Growing at ~25% per year over last several years |
| Most expensive AAV therapy (current) | Hemgenix / Beqvez — $3.5 million per dose (hemophilia B) |
| Zolgensma list price | $2.1 million (IV, pediatric SMA); Itvisma (intrathecal, older patients) priced at $2.59 million (approved November 2025) |
| Luxturna price | ~$850,000 (inherited retinal disease) |
| Zolgensma patients treated globally | Over 5,000 patients since 2019 approval |
| Zolgensma 2024 sales | $1.2 billion — world’s best-selling gene therapy |
| Neurological disorders segment share | 29.4–39% of AAV gene therapy market — largest therapeutic area |
| AAV9 clinical tissue focus | Ocular (26%), CNS (21%), liver (18%) of active trials |
| Primary route of administration | Intravenous (58%) of AAV therapy administrations |
| Key market players | Novartis (Zolgensma/Itvisma), Sarepta (Elevidys), Spark/Roche (Luxturna), CSL Behring (Hemgenix), Pfizer (Beqvez — discontinued), PTC Therapeutics (Kebilidi), BioMarin (Roctavian) |
| 2025 sector investment (H1) | $5 billion invested in cell and gene therapy sector in H1 2025 |
Source: Towards Healthcare (January 29 2026), Precedence Research (November 2025), IMARC Group, Roots Analysis (January 2026), Mordor Intelligence AAV CDMO (January 2026), BioInformant FDA-approved list (November 2025), PMC AAV gene therapy review (September 2025), Molecular Therapy (May 2025), PackGene H1 2025 analysis (November 2025), Drug Discovery News (October 2025), Fierce Pharma (November 2025)
The $2.75–3.85 billion market size figure for 2024–2025 might look modest for a technology described in sweeping terms — and it is modest, relative to the projected trajectory toward $112 billion by 2035. What that gap reflects is the fundamental structural challenge of gene therapy’s commercial model: these are treatments for rare and ultra-rare diseases with patient populations numbered in the hundreds or low thousands per indication in the US. Zolgensma treats roughly 450–500 new SMA births per year in the United States. Luxturna treats Leber congenital amaurosis, affecting approximately 1,000–2,000 Americans. Hemgenix targets hemophilia B, a population of roughly 33,000 patients in the US. The per-dose revenues of $2–3.5 million are built on health-economic arguments that a one-time curative intervention replaces decades of costly chronic therapy — and those arguments are largely correct, but they create reimbursement battles, access inequities, and payment structure challenges that are as consequential as the science itself.
The projected CAGR of 26–40% to 2034–2035 is driven by the assumption that the current rare-disease focus will expand into larger patient populations — heart failure, neurodegeneration, diabetes, cancer — where a single AAV treatment for a common condition could generate revenues dwarfing the entire current market. That expansion is scientifically plausible and is reflected in active trials, but it requires solving the immunogenicity problem (most adults have pre-existing AAV antibodies that block re-dosing), the manufacturing scale problem (current capacity cannot treat large patient populations at commercial prices), and the regulatory durability problem (long-term follow-up data for AAV therapies remains limited because the technology is still young). The North American 42–54% dominance reflects both the concentration of leading companies and the US’s relative advantage in having the world’s most developed market for ultra-high-cost specialty biologics.
FDA-Approved AAV Gene Therapies in the US | Complete Drug Profiles
| Product (Brand Name) | Generic Name | Indication | FDA Approval | Developer | Price / Dose | AAV Serotype |
|---|---|---|---|---|---|---|
| Luxturna | Voretigene neparvovec-rzyl | Leber congenital amaurosis / inherited retinal dystrophy (RPE65 mutation) | 2017 | Spark Therapeutics (Roche) | ~$850,000 | AAV2 |
| Zolgensma | Onasemnogene abeparvovec-xioi | Spinal muscular atrophy (SMA) — pediatric patients <2 years | 2019 | AveXis (Novartis) | $2.1 million | AAV9 |
| Roctavian | Valoctocogene roxaparvovec-rvox | Severe hemophilia A (adult patients) | 2023 (US) | BioMarin Pharmaceutical | Not disclosed (limited uptake) | AAV5 |
| Hemgenix | Etranacogene dezaparvovec-drlb | Hemophilia B (adult patients, FIX deficiency) | 2022 | CSL Behring | $3.5 million | AAV5 |
| Elevidys | Delandistrogene moxeparvovec-rokl | Duchenne muscular dystrophy (DMD) | 2023 (Accelerated Approval); expanded indication 2024 | Sarepta Therapeutics | Not disclosed | AAVrh74 |
| Beqvez | Fidanacogene elaparvovec | Hemophilia B (adult patients) | 2024 | Pfizer | $3.5 million | AAVRh74var |
| Kebilidi | Eladocagene exuparvovec-tneq | Aromatic L-amino acid decarboxylase (AADC) deficiency — first AAV therapy delivered directly into the brain | November 2024 | PTC Therapeutics | Not disclosed | AAV2 |
| Itvisma | Onasemnogene abeparvovec (intrathecal formulation) | SMA — older/heavier patients beyond pediatric IV indication | November 2025 | Novartis | $2.59 million | AAV9 |
Source: BioInformant FDA-approved cell and gene therapy list (November 2025), PMC viral vector gene therapies in the clinic (2025), Drug Discovery News (October 2025), Fierce Pharma (November 25 2025), Towards Healthcare AAV gene therapy market (January 2026)
The approval history of AAV gene therapies reads like a graduated proof of concept for the entire platform. Luxturna in 2017 was the breakthrough: a product targeting a tiny patient population (RPE65 mutation-driven retinal blindness) but demonstrating that a viral vector could restore meaningful function through a one-time intraocular injection, with durable results extending years beyond treatment. Its ~$850,000 price was unprecedented at the time and triggered the health-economic debates about gene therapy valuation that continue today. Zolgensma in 2019 was the paradigm-shifting moment: an AAV9-delivered SMN1 gene replacement that could prevent the progressive motor neuron death of SMA type 1 — the leading genetic cause of infant mortality — with a single IV infusion. Children who would have faced mechanical ventilation and early death instead walked, ran, and developed near-normally. The $2.1 million price tag was genuinely justified by health-economic modelling against lifetime chronic therapy costs, but it was also deeply controversial given that much of the foundational research was publicly funded.
The November 2025 approval of Itvisma — the intrathecal formulation of Zolgensma at $2.59 million — represents the most recent US AAV approval and a clinically important expansion: by delivering the vector directly into the spinal canal rather than intravenously, Itvisma can treat older and heavier SMA patients who cannot receive adequate doses via the IV route. Meanwhile, the discontinuation of Beqvez by Pfizer in early 2025 — despite FDA approval in 2024 — is a stark commercial cautionary tale. At a $3.5 million price point shared with Hemgenix, Beqvez found almost no patient uptake, largely because Hemgenix had already established itself in the hemophilia B market first. In a rare-disease space with tiny patient populations, being second to market for the same indication at the same price point is commercially untenable, and Pfizer’s retreat illustrates the brutal economics facing even genuinely efficacious AAV therapies.
AAV Gene Therapy by Disease & Serotype | US Therapeutic Data
| Category | Data | Source |
|---|---|---|
| Neurological disorders segment share | 29.4% of AAV gene therapy market (2024) — largest therapeutic area | Precedence Research (2025) |
| Neurological disorders driver | Alzheimer’s, Parkinson’s, SMA, rare neuropathies — AAV9 can cross blood-brain barrier | Towards Healthcare (2026) |
| Muscular disorders segment | Fastest-growing therapeutic area — driven by DMD, limb-girdle muscular dystrophy pipeline | Precedence Research (2025) |
| Hematologic disorders CAGR | Fastest CAGR of 32% in the AAV gene therapy market | Towards Healthcare (2026) |
| Ophthalmic disorders CAGR | 47% — significant growth rate through forecast period | Roots Analysis (2026) |
| Muscle-related disorders (AAV vector market) | 53% share of AAV vector market in 2026 — led by SMA and DMD | Roots Analysis (2026) |
| AAV9 serotype dominance | Largest serotype: 27.60–43% of AAV gene therapy market | Precedence Research / Towards Healthcare |
| Engineered capsids CAGR | 36% — fastest-growing serotype category | Towards Healthcare (2026) |
| Primary clinical tissue targets | Eye (26%), CNS (21%), liver (18%) — consistent across recent years | PackGene / H1 2025 analysis |
| Intravenous route dominance | 58% of AAV therapy administrations | Towards Healthcare (2026) |
| SMA US annual new diagnoses | ~450–500 newborns per year | BioPharma Dive (2019) / PMC Zolgensma review |
| SMA US death rate reduction | Zolgensma helped reduce US SMA infant mortality by two-thirds | ProPublica (2025) |
| SMA Type 1 survival (pre-treatment) | Less than 10% alive and off ventilation at age 2 without treatment | Novartis natural history data |
| Zolgensma STR1VE trial result | 21/21 patients alive without permanent ventilation; 47.6% achieved motor milestones (head control, sitting) | AJMC / FDA approval summary |
| Roctavian 7-year follow-up | High-dose cohort maintained mean FVIII activity of 16.2 IU/dL (mild hemophilia levels) at 7 years | PMC hemophilia gene therapy review (2025) |
| Hemgenix 5-year durability | FIX activity >30 IU/dL with minimal bleeding events over 5 years | PMC hemophilia gene therapy review (2025) |
| Elevidys patients treated (Jan 2025) | ~800 patients treated (clinical trials + commercial) | Molecular Therapy (May 2025) |
| Elevidys first patient death | March 2025 — 16-year-old non-ambulatory male, acute liver failure ~9 weeks post gene transfer | Molecular Therapy (May 2025) |
| Elevidys FDA hold | July 2025: FDA requested full pause; later lifted for ambulatory patients | PMC AAV review (September 2025) |
| Kebilidi significance | First AAV gene therapy delivered directly into the brain (intraparenchymal injection) for AADC deficiency | Towards Healthcare / Drug Discovery News |
| Hemophilia B US patient population | ~33,000 patients with hemophilia B in the US | General hematology data |
| Luxturna indication prevalence | ~1,000–2,000 Americans affected by RPE65-mutation retinal dystrophy | General ophthalmology data |
Source: Precedence Research (November 2025), Towards Healthcare (January 2026), Roots Analysis (January 2026), PackGene H1 2025 analysis (November 2025), Molecular Therapy (May 2025), PMC AAV-based gene therapy (September 2025), PMC hemophilia gene therapy review (2025), AJMC Zolgensma approval, ProPublica (June 2025), BioPharma Dive
The therapeutic target distribution in AAV gene therapy reflects both the biology of the vector and the commercial logic of rare disease development. AAV9’s ability to cross the blood-brain barrier makes CNS disorders a natural priority — and neurological conditions represent the largest single therapeutic segment at ~29–39% of market share. The genetic architecture of neurological diseases like SMA is relatively simple (single-gene loss of function) and well-characterized, making them scientifically tractable targets where functional proof of concept is achievable. The eye is an even more concentrated opportunity: ocular gene therapy benefits from immune privilege (the eye has limited immune surveillance compared to systemic tissues), allowing for direct subretinal injection with minimal immune response — exactly the biology that made Luxturna’s development feasible and its clinical results durable.
The hematology segment’s 32% projected CAGR reflects both the genuine clinical success of hemophilia gene therapies and the pipeline of next-generation programs targeting other blood disorders. However, the hemophilia commercial experience contains an important warning. Hemgenix’s $3.5 million price tag — the most expensive drug in the world at launch — drove intense payer pushback and very slow patient uptake. Pfizer’s Beqvez was essentially abandoned by its developer despite working as designed, because it arrived second to market for the same indication. The lesson is that scientific efficacy and commercial viability are not the same thing in gene therapy, and the field is learning that lesson in real time across multiple indications.
AAV Gene Therapy Challenges, Safety & Pipeline in the US | 2025–2026 Data
| Challenge / Development Area | Data / Status | Source |
|---|---|---|
| Immunogenicity — pre-existing antibodies | Significant portion of population carries pre-existing neutralizing antibodies to AAV serotypes — excludes patients from trials | Drug Discovery News / Pharma Almanac (2025) |
| Immunogenicity — immune response post-treatment | Delayed immune responses can occur; most AAV therapies are limited to a single lifetime administration | Pharma Almanac (2025) |
| Sarepta 2025 patient deaths | 3 patients with muscular dystrophy died of acute liver failure following AAV gene therapies in 2025 — all Sarepta clinical trials placed on hold | Drug Discovery News (October 2025) |
| Elevidys hold and resumption | FDA requested full pause July 2025; hold lifted for ambulatory patients — non-ambulatory patients remain restricted | PMC Molecular Therapy (2025) |
| Biogen AAV exit | September 2025: Biogen officially discontinued all AAV gene therapy programs | Drug Discovery News (October 2025) |
| Vertex Pharmaceuticals exit | April 2025: Vertex discontinued all internal AAV gene therapy research | Pharma Almanac (May 2025) |
| Pfizer Beqvez withdrawal | Early 2025: Pfizer discontinued global development of Beqvez — approved but minimal patient uptake; $3.5M pricing with no market differentiation | PMC viral vector review (2025) |
| Manufacturing challenges | Low yields; “empty capsids” (shells with no DNA) increase immunogenicity; scale-up from HEK293 or Sf9 systems remains challenging | Frontiers Molecular Medicine (December 2025) |
| Empty capsid standard (2025) | FDA now mandates orthogonal methods (two technologies) to quantify full vs. empty capsid ratio; mass photometry established as new gold standard in mid-2025 | BLA Regulatory / USP <1067> (2025) |
| AAV genome packaging limit | ~4.7–4.8 kb — prevents delivery of large genes (e.g., full-length dystrophin, CFTR) without engineering strategies (dual vectors, mini-genes) | PMC AAV review (September 2025) |
| Insertional mutagenesis risk | While rare, AAV can occasionally integrate into the genome — regulatory agencies now require more evidence on long-term integration safety | PMC AAV review (September 2025) |
| Liver toxicity concerns | Hepatotoxicity and thrombotic microangiopathy reported at high doses — particularly relevant for systemic delivery in large patients | Signal Transduction review (Nature, 2024) |
| USP reference standards launch | April 2025: US Pharmacopeia launched AAV reference standards and resources for developers and manufacturers | Precedence Research / Towards Healthcare |
| Novartis $1.1B Kate Therapeutics acquisition | November 2024: Novartis acquired Kate Therapeutics to improve AAV vector delivery technology | Drug Discovery News / Cell & Gene (2025) |
| Global CGT investment H1 2025 | $5 billion in H1 2025 — though start-up funding has slowed | PackGene H1 2025 |
| AAV CDMO market (2026) | $760 million — growing at 20.44% CAGR through 2031 | Mordor Intelligence (January 2026) |
| AAV CDMO market (2031) | $1.92 billion | Mordor Intelligence (January 2026) |
| Gene regulation therapies CAGR | 61% — fastest-growing application category within AAV vector market | Roots Analysis (2026) |
| AAV manufacturing service market (2026) | $1.66 billion — growing at 15.84% CAGR through 2035 | Towards Healthcare (November 2025) |
| Novartis Itvisma peak sales projection | Multibillion-dollar peak sales projected for intrathecal SMA therapy | Fierce Pharma (November 2025) |
| Sanofi SAR446268 fast track | September 2025: FDA granted fast-track designation for DM1 gene therapy — Sanofi candidate | Drug Discovery News (October 2025) |
| Rocket Pharmaceuticals RP-A501 hold lifted | August 2025: FDA lifted clinical hold on pivotal Phase 2 trial for Danon disease with optimized dosing | Drug Discovery News (October 2025) |
| Next-gen capsid engineering | Engineered capsids developed for improved tissue targeting, lower immunogenicity, and potential re-dosing | Pharma Almanac (2025) |
| LNP competition emerging | Lipid nanoparticles (LNPs) emerging as alternative to AAVs — re-dosable, no preexisting immunity concern, more scalable | Drug Discovery News (October 2025) |
| 2025 total US CGT clinical trials | North America: 844 active trials (of 1,905 globally) | PackGene / ARM / ASGCT H1 2025 |
Source: Drug Discovery News (October 2025), Pharma Almanac (May 2025), Frontiers Molecular Medicine (December 2025), PMC AAV-Based Gene Therapy (September 2025), BLA Regulatory (January 2026), Mordor Intelligence AAV CDMO (January 2026), Towards Healthcare AAV Manufacturing (November 2025), Cell & Gene 2025 Forecast (January 2025), PackGene H1 2025 (November 2025), PMC Viral Vector Gene Therapies in the Clinic (2025), Molecular Therapy (May 2025)
The 2025 exits of Biogen, Vertex, and (effectively) Pfizer from AAV gene therapy are being misread in much of the popular press as a collapse of confidence in the technology. The reality is more nuanced: these are strategic portfolio decisions by large, diversified pharmaceutical companies who concluded that the risk–reward profile of AAV-based programs was less favorable than other modalities — not a scientific verdict that AAV doesn’t work. Biogen’s exit followed years of disappointing results in neurological gene therapy, where the complexity of CNS targets and the immunogenicity challenges of high-dose systemic delivery have proven harder to manage than initially hoped. Vertex exited after concluding that CRISPR-based and mRNA-based approaches offered better profiles for its target indications. These decisions free capital for better-positioned programs and represent the natural selection of a maturing field rather than its obituary.
What is genuinely concerning — and demands serious scientific and regulatory attention — is the Sarepta Elevidys safety signal. Three deaths related to AAV gene therapies for muscular dystrophy in a single year is not a statistical blip in a treatment population of ~800 patients. The first documented death involved acute liver failure approximately nine weeks post-treatment in a non-ambulatory 16-year-old patient; additional cardiac-related deaths between 2 and 5 years post-vector have been noted, though causality assessments are ongoing. The FDA’s decision to lift the hold for ambulatory patients while maintaining restrictions for non-ambulatory patients reflects a careful risk stratification: the benefit–risk calculation differs meaningfully by patient ambulatory status and comorbidity profile. The broader lesson is one that the AAV field has known in principle but is now learning in clinical practice: immunogenicity and toxicity at high vector doses are not merely theoretical risks, and the innate immune response to capsid proteins can be catastrophic in vulnerable patients, particularly those with pre-existing cardiac compromise or advanced disease.
AAV Gene Therapy Manufacturing & Regulatory Landscape in the US | 2025–2026
| Manufacturing / Regulatory Area | Data / Status | Source |
|---|---|---|
| Global active cell & gene therapy candidates (2024) | Over 2,100 cell and gene therapy candidates in active development worldwide | Mordor Intelligence CDMO (2026) |
| Global gene therapies in clinical evaluation (2025) | More than 2,000 gene therapies under evaluation in various clinical phases | Roots Analysis (January 2026) |
| AAV clinical trial growth | Number of newly initiated AAV clinical trials peaked in 2023 and remained elevated through 2024 | PMC Viral Vector Gene Therapies (2025) |
| Primary AAV production systems | HEK293 cells (transient transfection): Luxturna, Zolgensma, Elevidys, Beqvez, Kebilidi; Sf9/baculovirus: Hemgenix, Roctavian | PMC AAV Gene Therapy (September 2025) |
| Suspension vs. adherent culture | Suspension HEK293 or Sf9 cultures dominate GMP production — more scalable than adherent systems | Frontiers Molecular Medicine (December 2025) |
| Empty capsid problem | Only ~7% of cells in a triple transfection model produce measurable assembled AAV capsids; empty capsids increase immunogenicity | Insights.bio AAV production article |
| AAV yield industry standard | Average yield of 3 × 10¹⁴ vg/L with 25% recovery — manufacturing bottleneck for large patient populations | Insights.bio (July 2024) |
| USP <1067> update (2025) | US Pharmacopeia recognized mass photometry in General Chapter as key orthogonal method for AAV characterization — mid-2025 | BLA Regulatory (January 2026) |
| USP AAV reference standards | April 2025: USP launched comprehensive package of AAV reference standards for developers and manufacturers | Precedence Research (2025) |
| FDA manufacturing flexibility (Phase 1) | FDA guidance: manufacturers NOT expected to fully comply with 21 CFR Part 211 prior to Phase 2/3 — allows capital savings in early stages | BLA Regulatory (January 2026) |
| Rapid sterility testing innovation | FDA accepting rapid PCR-based sterility and mycoplasma tests (e.g., BioFire) if validated — reduces release time from 28 days to hours | BLA Regulatory (January 2026) |
| Royalty burden on AAV patents | Royalties on foundational AAV patents can reach 25% of product revenue — eroding margins for price-sensitive indications | Mordor Intelligence CDMO (2026) |
| Biotech start-up CDMO demand | 49.08% of AAV CDMO market demand in 2025 came from biotech start-ups | Mordor Intelligence CDMO (2026) |
| Large pharma CDMO trajectory | Large pharma shows fastest CAGR at 20.81% as it internalizes gene therapy manufacturing platforms | Mordor Intelligence CDMO (2026) |
| Gene therapy revenue from AAV (gene market) | AAV represents 22% of total gene therapy market revenue (2023) | Grand View Research |
| FDA RMAT designation | Regenerative Medicine Advanced Therapy designation — accelerates review of promising gene therapies | General gene therapy regulatory |
| FDA platform technology designation | Sarepta’s viral vector for muscular dystrophy received FDA platform technology designation in 2025 | PackGene H1 2025 (November 2025) |
| Key CDMO players | Oxford Biomedica, Paragon Bioservices, GenScript Biotech, MeiraGTx, Lonza Group, Catalent, Thermo Fisher Scientific | Towards Healthcare manufacturing (November 2025) |
| GMP drug-substance suite revenue share | 38.05% of AAV CDMO revenue in 2025 | Mordor Intelligence CDMO (2026) |
| Downstream manufacturing growth | Fastest-growing CDMO service type over forecast period | Towards Healthcare manufacturing (2025) |
| China export halt (June 2025) | June 18, 2025: FDA announced immediate halt to new clinical trials involving export of US patient biological samples to “hostile countries” (including China) for genetic engineering — creating scrutiny for AAV supply chains with Chinese manufacturing | BLA Regulatory (January 2026) |
| FDA 10–20 annual approvals projection | FDA has projected 10–20 new cell and gene therapy approvals per year by 2025 | Grand View Research / multiple |
Source: Mordor Intelligence AAV CDMO (January 2026), Frontiers Molecular Medicine (December 2025), BLA Regulatory (January 2026), Towards Healthcare AAV Manufacturing (November 2025), PMC AAV gene therapy (September 2025), Insights.bio AAV production (July 2024), PackGene H1 2025 (November 2025), Grand View Research gene therapy market, Precedence Research (2025)
The manufacturing side of AAV gene therapy is where the distance between scientific promise and commercial reality is most painfully apparent. The core problem is one of scale: AAV vectors must be produced in enormous quantities of viral genome copies — doses for systemic delivery are measured in 10¹⁴ viral genomes per kilogram of patient body weight — yet current production platforms yield approximately 3 × 10¹⁴ viral genome copies per liter of bioreactor volume, with only 25% recovery after purification. A single dose of Zolgensma for a typical infant patient weighing 8 kg requires approximately 1.1 × 10¹⁴ vg/kg — meaning the manufacturing challenge is manageable for a very small patient. But scaling AAV production for conditions affecting hundreds of thousands or millions of patients (heart failure, Parkinson’s disease, Alzheimer’s disease) would require bioreactor capacity that does not currently exist at any commercially viable cost structure. This is not a theoretical future problem — it is actively constraining which indications are commercially viable targets today.
The empty capsid problem is one of manufacturing’s most insidious technical challenges. In typical AAV production, a significant proportion of the assembled viral particles contain no therapeutic DNA — they are empty shells with the correct protein coat but nothing functional inside. These empty capsids are immunogenic (they trigger immune responses just like full capsids) without providing any therapeutic benefit, meaning they contribute to immune toxicity while diluting the effective dose. The FDA’s 2025 mandate requiring orthogonal methods — two independent measurement technologies — to quantify full versus empty capsid ratios, and the USP’s recognition of mass photometry as a new gold standard for this measurement, represents a meaningful regulatory tightening that will improve product quality but also raise the bar for manufacturing compliance. The USP reference standards package launched in April 2025 similarly reflects a field-wide effort to create the analytical infrastructure that reproducible, scalable AAV manufacturing requires.
The geopolitical dimension of AAV manufacturing emerged sharply in 2025 with the FDA’s June 18 halt on exporting US patient biological samples to China for genetic engineering work. While explicitly targeting ex vivo cell therapies, the policy creates supply chain scrutiny for any AAV program with Chinese manufacturing partners — a meaningful operational concern given how much of the world’s plasmid production (a key starting material for AAV manufacturing) has been concentrated in Chinese contract facilities over the past decade. For developers who relied on Chinese CDMOs for cost efficiency, this represents both a regulatory risk and a supply chain vulnerability that is now prompting expensive and time-consuming domestic or European manufacturing shifts.
Disclaimer: The data reports published on The Global Files are sourced from publicly available materials considered reliable. While efforts are made to ensure accuracy, no guarantees are provided regarding completeness or reliability. The Global Files is not liable for any errors, omissions, or damages resulting from the use of these reports.